- HOME
- Products & Services
- Application Selection Guide
- Microbial genome sequencing
Microbial genome sequencing
Overview
Our service determines the genome sequences of microorganisms, including bacteria and fungi. You can choose between two sequencing methods: long-read sequencing and short-read sequencing.
・ Long-Read Sequencing
Recommended for constructing long contigs. This method requires high-molecular-weight DNA (>20 kb) and a larger quantity of genomic DNA compared to short-read sequencing.
Recommended for constructing long contigs. This method requires high-molecular-weight DNA (>20 kb) and a larger quantity of genomic DNA compared to short-read sequencing.
・ Short-Read Sequencing
Ideal for creating draft genomes at a lower cost or for samples where obtaining large amounts of genomic DNA is challenging.
Ideal for creating draft genomes at a lower cost or for samples where obtaining large amounts of genomic DNA is challenging.
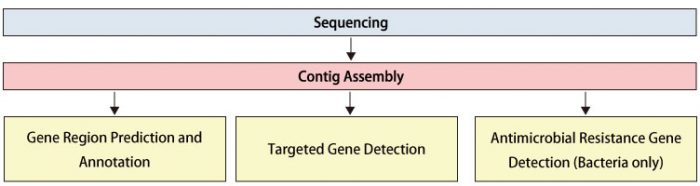
Workflow
Data Analysis
De Novo Assembly:
We assemble reads into contigs. Refer to FAQ section for details on the performance of assemblies using short-read and long-read sequencing.
We assemble reads into contigs. Refer to FAQ section for details on the performance of assemblies using short-read and long-read sequencing.
Example of Delivered Data: Contig Sequences (FASTA format)
>scaffold1
TTATCAAGAATACTTCGGTTCAATAAAAATGAATCCTGTGGAACTACTCCTAAATTTTGA
TTGATTAATATTTACACCATCAAATAAAATTTTTCCTGTATCAATCTTATATAATCCTGA
…
>scaffold2
AACATTCTCTTCATTTTCAAGCCAGATATCATTTACTCTATTTAAGTATCCTGCTGCCAA
TAAATATTGAGATTTCTGAACTAAATAAAATTGATGCCATGGTTTGAAATGCGACAACCT
…
TTATCAAGAATACTTCGGTTCAATAAAAATGAATCCTGTGGAACTACTCCTAAATTTTGA
TTGATTAATATTTACACCATCAAATAAAATTTTTCCTGTATCAATCTTATATAATCCTGA
…
>scaffold2
AACATTCTCTTCATTTTCAAGCCAGATATCATTTACTCTATTTAAGTATCCTGCTGCCAA
TAAATATTGAGATTTCTGAACTAAATAAAATTGATGCCATGGTTTGAAATGCGACAACCT
…
Base Correction Using Illumina Reads:
After assembling contigs with PacBio long-reads, we perform base correction using Illumina short-reads. Refer to FAQ section for details on the necessity of base correction.
After assembling contigs with PacBio long-reads, we perform base correction using Illumina short-reads. Refer to FAQ section for details on the necessity of base correction.
Gene Region Prediction and Annotation:
We predict ORF regions from the assembled contig sequences and assign annotation information through homology searches against known gene databases.
We predict ORF regions from the assembled contig sequences and assign annotation information through homology searches against known gene databases.
Example of Delivered Data: Annotation Results (EXCEL format)
| Position | Strand | Gene Name | Gene Product | Nucleotide Sequence | Amino Acid Sequence |
|---|---|---|---|---|---|
| sequence01:218-1240 | – | pcpC | choline-binding protein F | ATGAAGCTTTTGAAAAA… | MKLLKKMMQVALATFFFG… |
| sequence01:3331-3963 | – | rr03 | DNA-binding response regulator | ATGAAAATTTTACTAGT… | MKILLVDDHEMVRLGLKS… |
| sequence01:3977-4972 | – | hk03 | sensor histidine kinase | ATGAAAAAACAAGCCTA… | MKKQAYVIIALTSFLFVF… |
| sequence01:5744-6754 | – | fni | isopentenyl-diphosphate delta-isomerase | ATGACGACAAATCGTAA… | MTTNRKDEHILYALEQKS… |
| sequence01:6738-7745 | – | mvaK2 | phosphomevalonate kinase | ATGATTGCTGTTAAAAC… | MIAVKTCGKLYWAGEYAI… |
Example of Delivered Data: DDBJ Submission File (Bacteria only)
| sequence01 | source | 1..278302 | mol_type | genomic DNA |
| organism | Streptococcus pneumoniae | |||
| submitter_seqid | @@[entry]@@ | |||
| ff_definition | @@[organism]@@ @@[strain]@@ DNA, @@[submitter_seqid]@@ | |||
| CDS | complement(218..1240) | product | choline-binding protein F | |
| codon_start | 1 | |||
| inference | COORDINATES:ab initio prediction:MetaGeneAnnotator | |||
| inference | similar to AA sequence:RefSeq:WP_000771073.1 | |||
| gene | pcpC | |||
| locus_tag | LOCUS_00010 | |||
| transl_table | 11 |
Antimicrobial Resistance Gene Detection (Bacteria only):
We detect antimicrobial resistance genes within bacterial genomes.
Example of Delivered Data: Antimicrobial Resistance Gene Detection Results (EXCEL format)
| Position | Strand | Gene Name | Homology (%) | Resistance |
|---|---|---|---|---|
| chromosome1:38381-39151 | + | ant(4′)-Ia | 99.87 | KANAMYCIN/TOBRAMYCIN |
| chromosome1:39368-39772 | + | bleO | 100 | BLEOMYCIN |
| chromosome1:44969-46975 | – | mecA | 100 | METHICILLIN |
| chromosome1:47075-48049 | + | mecR1 | 100 | METHICILLIN |
| chromosome1:126289-127641 | + | tet(38) | 100 | TETRACYCLINE |
| chromosome1:2479070-2479489 | + | fosB-Saur | 99.76 | FOSFOMYCIN |
Sample Requirements
| Method | Library Preparation | Sample Type | Total Amount | Concentration | Volume |
|---|---|---|---|---|---|
| Long-read sequencing | – | High molecular weight DNA | ≥ 10 µg | ≥ 50 ng/µL | ≥ 30 µL |
| Short-read sequencing | PCR-Plus | DNA | ≥ 500 ng | ≥ 10 ng/µL | ≥ 30 µL |
| PCR-Free | ≥ 4 µg | ≥ 50 ng/µL | ≥ 30 µL |
Frequently Asked Questions
Q: What are the differences in assembly results between long-read and short-read sequencing?
Short-read sequencing struggles with repeat regions, often resulting in fragmented contigs. Long-read sequencing can capture repeat regions, producing longer contigs during assembly.
Assembly Results
| * The above are reference values based on our actual performance. | |||
| Organism Type | Genome Size | Number of Contigs | |
|---|---|---|---|
| Long-read sequencing | Short-read sequencing | ||
| Bacteria | 1 – 10 Mb | 1 – 5 | 30 – 300 |
| Fungi | 10 – 100 Mb | 20 – 400 | 200 – 4,000 |
Q: Can complete bacterial genomes be generated using long-read sequencing?
Using PacBio reads, most bacterial genomes in our experience assemble into a single contig. However, genome structures or sample conditions may occasionally result in multiple contigs.
Q: Is base correction necessary after assembly with PacBio reads?
The contig sequences constructed from PacBio reads may contain systematic base calling errors, such as homopolymer misreads (e.g., AAAA misinterpreted as AAA). These insertions or deletions (InDels) can potentially impact gene prediction frames. To ensure accurate annotation, we recommend performing base corrections prior to annotation.
Q: How can fragmented contigs be scaffolded?
If a complete genome sequence of a closely related strain is available, it can serve as a reference for scaffolding contigs. However, differences in genome structure between the reference and sample may result in errors, so we recommend verifying sequences using capillary sequencing.