- HOME
- Products & Services
- Application Selection Guide
- Small RNA-Seq
-
Oligonucleotide API CRDMO
- Oligonucleotide API CRDMO – Service Overview
- Antisense Oligonucleotide (ASO) Design & Synthesis
- RUO Oligonucleotide Synthesis (in vitro / in vivo)
- Evaluation of Off-target Effects
- Cell-Based Assays & Screening
- Animal Experimentation & Non-Clinical Trial
- GMP Oligonucleotide APIs: Manufacturing & Quality Control
-
Custom DNA/RNA Synthesis
-
Next Generation Sequencing
- Next Generation Sequencing
-
Application Selection Guide
- Application Selection Guide
- Human Genome Sequencing
- Whole Genome Sequencing (Non-Human)
- Microbial genome sequencing
- Small scale sequencing (NGS Petit)
- GRAS-Di® Genotyping
- Repeat Motif Detection
- Library Prep for Challenging DNA Samples
- Gene Expression Analysis (Reference-Based)
- Isoform Sequencing (full-length mRNA-seq)
- De novo Transcriptome Sequencing
- Small RNA-Seq
- Microbial Community Analysis
- Shotgun Metagenomic Sequencing
- Metatranscriptome Sequencing
- ChIP-Seq
- CRISPR Screening
- Illumina Amplicon Sequencing
- PacBio Amplicon Sequencing
- Standard Pipeline Data Analysis
- Custom Data Analysis
- Gene Analysis from Pathological Specimens
- Sequencer Models and Sequencing Principles
- Sample Requirements
- How to Order
- Scientific Publications
-
Custom DNA Sequencing
-
Custom DNA Microarray
-
Protein Related Service
-
Laboratory Tools and Service
Small RNA-Seq
Overview
This service analyzes the expression of small RNAs, such as miRNAs, using Small RNA-Seq data.
Workflow
Specifications
| Platform | Illumina NovaSeq |
| Data Amount | 10 million read pairs per sample |
Data Analysis
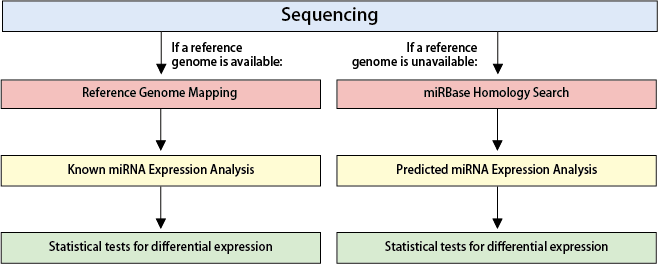
【Reference Genome Mapping and Known miRNA Expression Analysis】
Reads from Small RNA-Seq data are mapped to the reference genome to quantify the expression levels of known miRNAs. The standard analysis includes calculating Fold Changes between samples or groups.
Reads from Small RNA-Seq data are mapped to the reference genome to quantify the expression levels of known miRNAs. The standard analysis includes calculating Fold Changes between samples or groups.
【miRBase Homology Search and Predicted miRNA Expression Analysis】
Reads are clustered into identical sequences and compared against known miRNAs in miRBase. Expression levels of predicted miRNAs are estimated based on read counts, followed by inter-sample or inter-group comparisons to calculate Fold Changes.
Reads are clustered into identical sequences and compared against known miRNAs in miRBase. Expression levels of predicted miRNAs are estimated based on read counts, followed by inter-sample or inter-group comparisons to calculate Fold Changes.
【Statistical Testing】
Differential expression analysis for miRNAs is conducted using statistical tests. Replicates of n≥3 are recommended for robust analysis.
Differential expression analysis for miRNAs is conducted using statistical tests. Replicates of n≥3 are recommended for robust analysis.
Sample Requirements
| Sample Type | Total Amount | Concentration | Volume |
| Total RNA | ≥ 2 µg | ≥ 50 ng/µL | ≥ 30 µL |
| Small RNA | ≥ 60 ng | ≥ 2 ng/µL | ≥ 30 µL |