- HOME
- Products & Services
- Application Selection Guide
- De novo Transcriptome Sequencing
De novo Transcriptome Sequencing
- Overview
- Workflow
- Recommended Data Amount
- Data Amount
- Sample Requirements
- Frequently Asked Questions
Overview
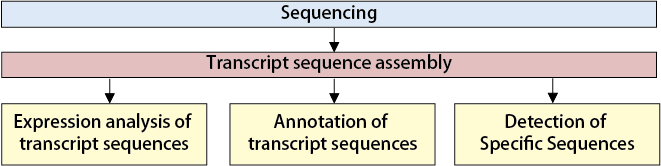
We provide a service to construct transcript sequences from RNA-Seq data. This enables gene detection and expression analysis for organisms without a reference genome.
Workflow
Recommended Data Amount
| Sequencing Specification | Illumina 150PE |
| Data Amount | 20 million read pairs per sample (See FAQ for details) |
Data Amount
【De novo Assembly】
Assembles reads to construct transcript sequences. For mixed-sample assembly, refer to the FAQ.
Assembles reads to construct transcript sequences. For mixed-sample assembly, refer to the FAQ.
Example of Delivered Data: Transcript sequences (FASTA format)
>TR1|c0_g1_i1
ATTTTTATTTTGATGTGAAAAGTTAATTTTTTAACTATAAAATATTTTGCTTCAACAAGA
CAATATCCGCCAAATTAAATGAATATGCATTTATTATCTTTACCTGATTGTATGCTGGAA
…>TR1|c0_g1_i2
CGCCGAGAATCCGAGAGGAAAAAATGTATTTTCTTTCAGAAATCATCCACTTGCGAAACA
TGCTGACATACTAACTTGTATTGAAACAAGAATATCCATGCTTTCAATGACATACACCAA
…
ATTTTTATTTTGATGTGAAAAGTTAATTTTTTAACTATAAAATATTTTGCTTCAACAAGA
CAATATCCGCCAAATTAAATGAATATGCATTTATTATCTTTACCTGATTGTATGCTGGAA
…>TR1|c0_g1_i2
CGCCGAGAATCCGAGAGGAAAAAATGTATTTTCTTTCAGAAATCATCCACTTGCGAAACA
TGCTGACATACTAACTTGTATTGAAACAAGAATATCCATGCTTTCAATGACATACACCAA
…
【Transcriptome Annotation】
Performs BLAST searches using established gene databases to annotate transcript sequences generated by assembly.
Performs BLAST searches using established gene databases to annotate transcript sequences generated by assembly.
【Mapping and Expression Analysis】
Maps original reads to the assembled transcript sequences and quantifies gene expression levels. Comparisons between samples or groups are also supported.
Maps original reads to the assembled transcript sequences and quantifies gene expression levels. Comparisons between samples or groups are also supported.
Example of Delivered Data: Expression analysis results (EXCEL format)
| gene_id | Sample A | Sample B | Sample B | |||
| Sample A | ||||||
| FPKM | FPKM | logFC | logCPM | PValue | FDR | |
| TR10000|c0_g1 | 0.49 | 0.13 | -1.824253682 | -2.050135117 | 0.430641822 | 1 |
| TR10001|c0_g1 | 0.13 | 0.7 | 2.238008044 | -1.894306859 | 0.282608696 | 0.908307924 |
| TR10002|c0_g1 | 0.58 | 0.25 | -1.218299202 | -1.760545939 | 0.52173913 | 1 |
| TR10003|c0_g1 | 0.08 | 0.77 | 3.070569548 | -1.407003958 | 0.057771114 | 0.475628079 |
| TR10004|c0_g1 | 0.5 | 0.77 | 0.52824399 | -1.217384592 | 0.82249198 | 1 |
| TR10005|c0_g1 | 20.61 | 16.37 | -0.408638264 | 3.253900296 | 0.555121307 | 1 |
| … | … | … | … | … | … | … |
【Detection of Specific Sequences】
Searches for sequences homologous to specified ones* within the assembled transcript sequences.
Searches for sequences homologous to specified ones* within the assembled transcript sequences.
* Example: Known peptide fragments or genes from closely related species
Sample Requirements
| Organism Type | Sample Type | Total Amount | Concentration | Volume |
| Eukaryotes | Total RNA | ≥ 600 ng | ≥ 20 ng/µL | ≥ 30 µL |
| mRNA | ≥ 100 ng | ≥ 3 ng/µL | ≥ 30 µL | |
| Prokaryotes | Total RNA | ≥ 1.5 µg | ≥ 50 ng/µL | ≥ 30 µL |
| mRNA | ≥ 100 ng | ≥ 3 ng/µL | ≥ 30 µL |
Frequently Asked Questions
Q: Recommended Data Amount
For assembly using data from a single sample, we recommend at least 50 million read pairs per sample. For mixed-sample assembly, analysis is possible with 10-20 million read pairs per sample. Increasing the data amount enables the detection of lower-expressed genes.
For assembly using data from a single sample, we recommend at least 50 million read pairs per sample. For mixed-sample assembly, analysis is possible with 10-20 million read pairs per sample. Increasing the data amount enables the detection of lower-expressed genes.
Q: Number and Length of Contigs from De novo Assembly
Depending on the organism and sequencing data amount, contigs typically average 800-1,000 bp in length, with a total count of around 100,000-300,000 contigs. For obtaining full-length transcript sequences, we recommend Iso-Seq analysis using a PacBio sequencer.
Depending on the organism and sequencing data amount, contigs typically average 800-1,000 bp in length, with a total count of around 100,000-300,000 contigs. For obtaining full-length transcript sequences, we recommend Iso-Seq analysis using a PacBio sequencer.
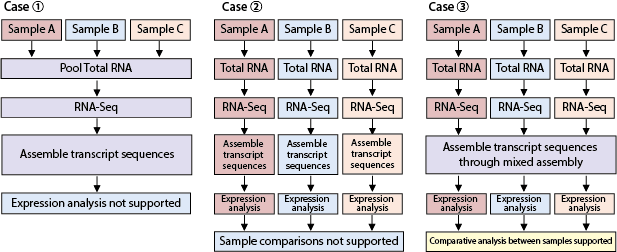
Q: Mixed Assembly
When the primary goal is obtaining gene sequence information from an organism, pooling Total RNA from various tissues ensures balanced gene expression profiles for sequencing and assembly (Case ①). For obtaining expression profiles of individual samples (Case ②) or performing comparative expression analysis between samples (Case ③), RNA-Seq data must be acquired for each sample.
When the primary goal is obtaining gene sequence information from an organism, pooling Total RNA from various tissues ensures balanced gene expression profiles for sequencing and assembly (Case ①). For obtaining expression profiles of individual samples (Case ②) or performing comparative expression analysis between samples (Case ③), RNA-Seq data must be acquired for each sample.
Q: Replicates for Expression Analysis
For statistical testing of differential expression, we recommend at least three replicates (n=3).
While analysis without replicates is possible, it will provide only fold-change information between samples.
For statistical testing of differential expression, we recommend at least three replicates (n=3).
While analysis without replicates is possible, it will provide only fold-change information between samples.
Q: Is De novo Transcriptome Analysis Feasible for Bacteria?
For bacteria, we recommend constructing a genome through genome sequencing and predicting genes directly from the genome, rather than performing De novo transcriptome analysis.
For bacteria, we recommend constructing a genome through genome sequencing and predicting genes directly from the genome, rather than performing De novo transcriptome analysis.