- HOME
- Products & Services
- Application Selection Guide
- Human Genome Sequencing
Human Genome Sequencing
- Overview
- Analysis Methods
- Workflow
- Recommended Data Amount
- Data Analysis
- Sample Requirements
- Frequently Asked Questions
Overview
Our human genome sequencing service identifies potential genetic variants associated with diseases and other conditions. Through comprehensive genome analysis, we help pinpoint candidate mutations efficiently.
Analysis Methods
Whole Genome Sequencing (WGS):
WGS includes introns and intergenic regions, making it suitable for detecting germline mutations. However, achieving high coverage across the entire genome can be challenging.
Exome Sequencing:
Exome sequencing focuses on coding regions, providing high coverage and enabling the detection of low-frequency mutations, such as those found in cancer tissues.
Workflow
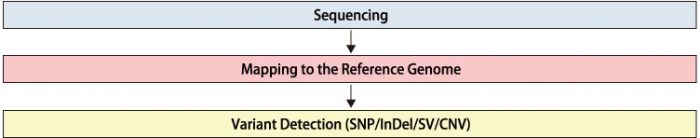
Recommended Data Amount
| Method | Target Region | Target Size | Example | |
| Data Amount | Coverage*2 | |||
| Whole Genome Sequencing | Entire Genome | 3 Gb | 90 Gb | x 30 |
| Exome Sequencing*1 | Entire Exome | 60 Mb | 6 Gb | x 50 |
*1 For exome sequencing, we use the Agilent SureSelect Human All Exon V6 kit.
*2 Coverage values are estimates accounting for capture efficiency and biases.
Data Analysis
Mapping and Variant Detection:
Sequencing reads are aligned to the reference genome to identify candidate SNPs, InDels, SVs, and CNVs.
Sequencing reads are aligned to the reference genome to identify candidate SNPs, InDels, SVs, and CNVs.
* SVs and CNVs are detectable only through Whole Genome Sequencing.
Sample Requirements
| Method | Target Region | Library Preparation | Sample Type | Total Amount | Concentration | Volume |
| Whole Genome Sequencing | Entire Genome | PCR-Plus | DNA | ≥ 500 ng | ≥ 10 ng/µL | ≥ 30 µL |
| PCR-Free | ≥ 4 µg | ≥ 50 ng/µL | ≥ 30 µL | |||
| Exome Sequencing | Entire Exome | PCR-Plus | ≥ 2 µg | ≥ 10 ng/µL | ≥ 30 µL |
Frequently Asked Questions
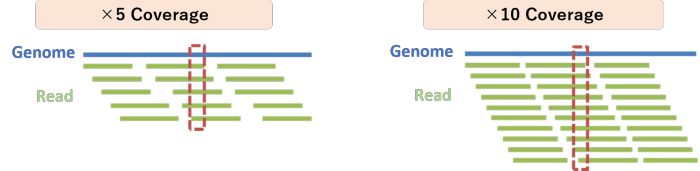
Q:What is coverage, and how does it relate to mutation frequency?
Coverage represents the average number of sequencing reads covering a position in the target region, calculated as:
Data Output (bp) ÷ Target Size (bp)
In exome sequencing, the actual coverage may be lower due to capture efficiency and bias.
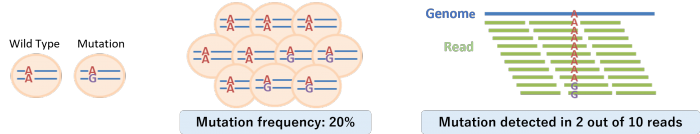
Mutation frequency refers to the proportion of cells in the sample carrying a specific mutation.
For example, a heterozygous mutation present in 40% of cancer cells corresponds to a mutation frequency of 20%.
Q:Are control samples necessary?
Performing mutation analysis with only one sample, regardless of disease status, may lead to the detection of many mutation candidates arising from individual genetic variation between the reference genome and the sample’s genome, as well as from misalignments (e.g., around 4 million in whole-genome human analysis, or about 500,000 in exome analysis).
For somatic mutations, matched normal tissue from the same individual is typically used as a control. For germline mutations, samples from close relatives are often used. These control samples help distinguish true mutations from background noise.
Performing mutation analysis with only one sample, regardless of disease status, may lead to the detection of many mutation candidates arising from individual genetic variation between the reference genome and the sample’s genome, as well as from misalignments (e.g., around 4 million in whole-genome human analysis, or about 500,000 in exome analysis).
For somatic mutations, matched normal tissue from the same individual is typically used as a control. For germline mutations, samples from close relatives are often used. These control samples help distinguish true mutations from background noise.
Q:How can I validate low-frequency mutations?
Validation can be performed by amplifying the mutation region through PCR and performing deep sequencing (see Amplicon Sequencing ).
Validation can be performed by amplifying the mutation region through PCR and performing deep sequencing (see Amplicon Sequencing ).