- HOME
- Products & Services
- Application Selection Guide
- Whole Genome Sequencing (Non-Human)
Whole Genome Sequencing (Non-Human)
- Overview
- Workflow
- Recommended Data Amount
- Data Analysis
- Sample Requirements
- Frequently Asked Questions
Overview
Genome sequencing is performed to identify potential mutations across the genome. This allows for the exploration of genes responsible for mutant strains.
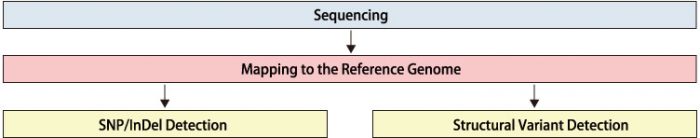
Workflow
Recommended Data Amount
| Sequencing Specification | Illumina 150PE |
| Data Amount | ≥ 30x of genome size. Please refer to the FAQ section for further details.
Example : Arabidopsis thaliana (Genome Size: 130Mb) → Recommended Data Amount: ≥ 4Gb |
Data Analysis
Mapping and SNP Candidate Detection:
Reads are mapped to the reference genome, and SNP candidate positions are identified by comparing the reference sequence with the read sequences.
Reads are mapped to the reference genome, and SNP candidate positions are identified by comparing the reference sequence with the read sequences.
Example of Delivered Data: Mutation Detection Results (Excel Format)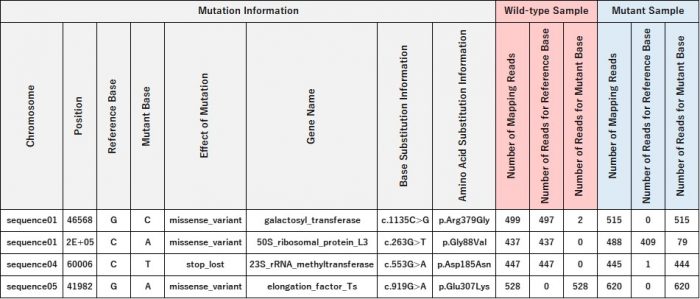
Mapping and Structural Variant Detection:
Reads are mapped to the reference genome, and based on paired-read mapping, large-scale structural variants (insertions, deletions, inversions, translocations) are identified.
Reads are mapped to the reference genome, and based on paired-read mapping, large-scale structural variants (insertions, deletions, inversions, translocations) are identified.
Sample Requirements
| Library Preparation | Sample Type | Total Amount | Concentration | Volume |
| PCR-Plus | DNA | ≥ 500 ng | ≥ 10 ng/µL | ≥ 30 µL |
| PCR-Free | ≥ 4 µg | ≥ 50 ng/µL | ≥ 30 µL |
Frequently Asked Questions
Q:Coverage and Data Amount?
Coverage represents the average number of sequencing reads covering any given position in the genome, calculated as:
Data Output (bp) ÷ Target Size (bp)
Coverage represents the average number of sequencing reads covering any given position in the genome, calculated as:
Data Output (bp) ÷ Target Size (bp)
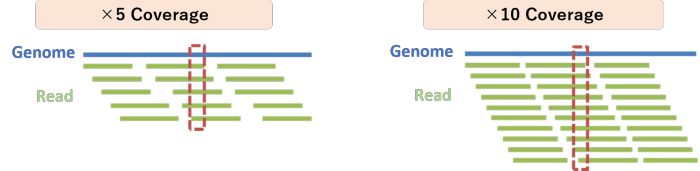
For example, when performing mutation analysis with data at an average 30x coverage, homozygous mutations are detected in nearly all of the 30 reads, while heterozygous mutations are detected in about 15 out of 30 reads.
Coverage may vary across different regions, so it is recommended to acquire data with at least 30x average coverage.
Coverage may vary across different regions, so it is recommended to acquire data with at least 30x average coverage.
Q:Are control samples necessary?
When mutation analysis is performed with only one mutant sample, individual genetic differences between the reference genome and the sample, as well as misalignments, can result in the detection of many mutation candidates.
By analyzing control samples, such as parental or wild-type strains, these background mutations can be excluded, allowing for the extraction of mutations specific to the target sample.
When mutation analysis is performed with only one mutant sample, individual genetic differences between the reference genome and the sample, as well as misalignments, can result in the detection of many mutation candidates.
By analyzing control samples, such as parental or wild-type strains, these background mutations can be excluded, allowing for the extraction of mutations specific to the target sample.
Q:Bulk Sample Analysis
In fungi and plants, mutations obtained through mutagenesis may include mutations unrelated to traits.
Analyzing bulk samples consisting of 10-30 mutants from backcrosses helps distinguish causative mutations from other mutations based on mutation frequency.
In this case, wild-type or parental strain bulk samples are used as controls (see “Are control samples necessary?“).
In fungi and plants, mutations obtained through mutagenesis may include mutations unrelated to traits.
Analyzing bulk samples consisting of 10-30 mutants from backcrosses helps distinguish causative mutations from other mutations based on mutation frequency.
In this case, wild-type or parental strain bulk samples are used as controls (see “Are control samples necessary?“).
Q:Microbial Genome Analysis
For small microbial genomes, if the sample’s strain is distantly related to the reference genome in public databases, we recommend constructing a reference genome using wild-type samples (see Microbial Genome Sequencing).
For small microbial genomes, if the sample’s strain is distantly related to the reference genome in public databases, we recommend constructing a reference genome using wild-type samples (see Microbial Genome Sequencing).
Q:Can RNA-Seq Data be Used for Mutation Detection?
RNA-Seq data can be used to detect for SNPs in genes that are sufficiently expressed. However, there are potential issues, such as the detection of false-positive mutations near splice junctions or for genes with low expression.
Additionally, if there is differential expression between alleles, accurate mutation frequency calculation across the genome can be problematic.
RNA-Seq data can be used to detect for SNPs in genes that are sufficiently expressed. However, there are potential issues, such as the detection of false-positive mutations near splice junctions or for genes with low expression.
Additionally, if there is differential expression between alleles, accurate mutation frequency calculation across the genome can be problematic.