- HOME
- Products & Services
- Sequencer Models and Sequencing Principles
Sequencer Models and Sequencing Principles
Next-Generation Sequencer Models
Illumima
| Sequencer | Run Condition | Estimated Reads per Lane | Read Length | Data Amount | Multiplexing Support |
| MiSeq | V2 | 20 million reads (10 million read pairs) |
150 PE | 3Gb | – |
| 250 PE | 5Gb | – | |||
| V3 | 32 million reads (16 million read pairs) |
75 PE | 2.5Gb | – | |
| 300 PE | 10Gb | Supported (in increments of 100,000 read pairs) |
|||
| NextSeq | MidOutput | 260 million reads (130 million read pairs) |
75 PE | 19.5Gb | – |
| 150 PE | 39Gb | – | |||
| HighOutput | 400 million reads | 75 PE | 30Gb | – | |
| 800 million reads (400 million read pairs) |
75 PE | 60Gb | – | ||
| 150 PE | 120Gb | – | |||
| NovaSeq | S4 | 5.2 billion reads (2.6 billion read pairs) |
150 PE | 800Gb | Supported (in 1 Gb increments) |
PacBio
| Sequencer | Supported Cells | Estimated Reads per Run | Read Length | Data Amount | Multiplexing Support |
| Sequel | 1M ZMW | Approx. 500,000 reads | 10~15kb | 1~3Gb | – |
| Sequel II / IIe | 8M ZMW | Approx. 4 million reads | 10~15kb | 15~30Gb | Supported * Sequel II / Revio model selection not available |
| Revio | 25M ZMW | Approx. 12 million reads | 10~15kb | 約50Gb |
Sequencing Principles
Illumina
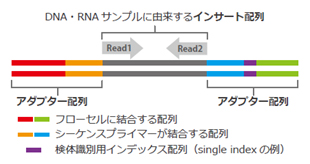
The Illumina sequencing library consists of insert sequences derived from DNA or RNA samples with adapters attached at both ends.
The library binds to the flow cell of the sequencer, where each individual molecule is amplified to form a cluster.
In each cluster, sequencing occurs in cycles of single-base extension followed by fluorescence detection.
| <Illumina Sequencing Library> | <Illumina Sequencing Principle> |
 |
 |
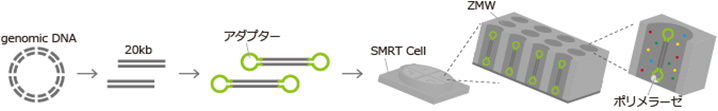
PacBio
The PacBio sequencing library consists of long insert sequences (10-20 kb) with hairpin adapters at both ends.
The SMRT Cell contains nanometer-sized wells called ZMWs, where single-molecule sequencing occurs.
In a single-molecule library, the polymerase continuously cycles through the template, sequencing tens of kilobases.
<PacBio Sequencing Principle> *Example: Genome Sequencing Analysis
 |
||
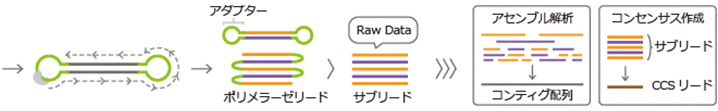
| Genomic DNA is physically fragmented, and fragments of ~20 kb are collected. | Hairpin adapters are ligated to both ends of the DNA fragments. | A single-molecule library binds with polymerase in ZMWs of the SMRT Cell, enabling real-time DNA synthesis detection. |
 |
||
| Circularized libraries are sequenced repeatedly. | Polymerase reads are generated, adapter sequences are removed, and subreads are obtained. | Subread data are used for further data analysis. |
Frequently Asked Questions
A FASTQ file is one of the raw data formats output by next-generation sequencers. It contains both read sequences and quality scores.
Each read’s information is represented in four lines:
– Line 1 & Line 3: Read-specific ID
– Line 2: Nucleotide sequence
– Line 4: Quality score
The quality score correlates with the Phred Score used in the Sanger sequencing method and is represented as a character corresponding to a numerical value between 0 and 42.
【 FASTQ format】

FASTQ files can be opened as text files. If compressed (e.g., GZ format), decompression is required before opening.
For large file sizes, specialized text viewer software may be necessary.
Raw data from paired-end sequencing is output as separate FASTQ files for Read 1 and Read 2. Typically, files are named as follows:
“[filename]_Read1/Read2.fastq” or “[filename]_R1/R2.fastq”
The paired reads appear in corresponding order in both files.
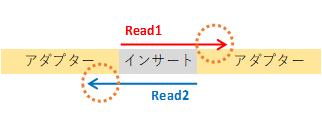
In Illumina sequencing, reads are generated from the ends of the insert sequence:
– Single-end sequencing: Only Read 1 is sequenced.
– Paired-end sequencing: Both Read 1 and Read 2 are sequenced.
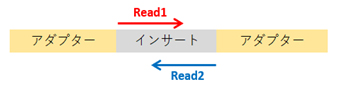
An index sequence is a short (6-8 bp) identifier embedded within the adapter.
Our raw data output includes index sequences within the sequence ID of FASTQ files.
A single index sequences the index within one side of the adapter, while a dual index sequences the indexes within both adapters.
Depending on the sequencer model and the version of the run reagents, the index read direction may vary.

If the insert sequence is shorter than the read length, adapter sequences may appear at the 3′ end of reads.
We recommend trimming the 3′ adapter sequences before analysis.
The 5′ end is sequenced directly downstream of the adapter and generally does not contain adapter sequences.
Our Illumina raw data undergoes chastity filtering, but no additional quality filtering is applied.
As sequencing accuracy may decline towards the latter part of reads, we recommend performing quality-based trimming or filtering as needed.
Illumina sequencers capture fluorescence images for each sequencing cycle (one-base extension per cycle).
The base with the highest signal intensity is recorded. Chastity filtering excludes unreliable reads based on the following formula:
For the first 25 cycles, if (I1 / (I1 + I2)) > 0.6 fails in two or more cycles, the read is discarded.
(I1 = highest signal intensity, I2 = second highest signal intensity)