- HOME
- Products & Services
- Guide to select the best application for your purpose
- Microbial community analysis
Microbial community analysis
★データ解析アップデート★
これまで有料オプション(追加解析)としていました下記の解析を標準解析メニューにてご提供いたします
解析概要
糞便、土壌などから抽出したゲノムDNAを対象として、16S rRNAなどのマーカー遺伝子のPCR増幅、ディープシーケンスを行い、サンプルに含まれる微生物の種類と占有率を明らかにします。
解析ワークフロー
| 土壌・糞便などからのゲノムDNA抽出 | ||||
| ↓ | ||||
| 1st PCR(ターゲット領域の増幅、リンカー配列付加) | ||||
| ↓ | ||||
| 2nd PCR(アダプター配列付加) | ||||
| ↓ | ||||
| シーケンスデータ取得 | ||||
| ↓ | ||||
| 標準データ解析(菌叢構成情報の算出、α多様性情報の算出) | ||||
| ↓ | ↓ | ↓ | ||
| PCoAプロット作成 | 有意差検定 (PERMANOVA) |
有意差検定 (Differential Abundance Analysis) |
||
解析対象領域(PCRプライマー)
弊社では下記のプライマーをご提供しています。
その他の解析対象領域については、ご相談ください。
| 解析対象領域 | プライマーの特徴 | 文献情報 |
|---|---|---|
| Bacteria 16S rRNA V3-V4 (341F / 805R) |
弊社で標準的に使用しているプライマーです。 真正細菌と、一部の古細菌を増幅します。 |
Herlemann, Daniel PR, et al. The ISME journal 5.10 (2011): 1571-1579. |
| Bacteria 16S rRNA V3-V4 (341F’ / 805R) |
上記「341F / 805R」よりも古細菌の増幅効率が高いプライマーセットです。 | Hugerth, Luisa W., et al. Applied and environmental microbiology 80.16 (2014): 5116-5123. |
| Bacteria 16S rRNA V3-V4 (335F / 769R) |
植物の葉緑体16S rRNAの増幅を抑え、細菌由来の16S rRNAを特異的に増幅します。植物が付着・混入したサンプルにお勧めです。 | Dorn-In, Samart, et al. Journal of microbiological methods 113 (2015): 50-56. |
| Fungi ITS1 (ITS1-F_KYO1 / ITS2_KYO2) |
真菌に対応したプライマーセットです。 | Toju, Hirokazu, et al. PloS one 7.7 (2012): e40863. |
解析プラン
<乗り合い解析>
シーケンスデータ量:MiSeq 300bp ペアエンド 約10万リードペア/1検体
| DNA抽出 | お客様にて実施 or 15,000円/検体※ |
|---|---|
| 1st PCR~精製 | お客様にて実施 or 10,000円/検体 |
| 2nd PCR~精製 | 60,000円/検体 |
| MiSeqシーケンスデータ取得 | |
| データ解析 |
<1RUNご購入>
シーケンスデータ量:
MiSeq 300bp ペアエンド 約1600万リードペア/1RUN(約30%はPhiXコントロール)
サンプル必要量
| サンプル | 必要量 | 保管・輸送温度 |
|---|---|---|
| 汚泥 | 5~10mL | 冷蔵 |
| 土壌 | 1~5g | 冷蔵 |
| 糞便 | 0.2~0.5g | 冷凍 |
| 唾液 | 0.2~1mL | 冷凍 |
| サンプル | 総量 | 濃度 | 液量 |
|---|---|---|---|
| 精製済みゲノムDNA | 150 ng 以上 | 5 ng/uL 以上 | 30 uL 以上 |
| サンプル | 総量 | 濃度 | 液量 |
|---|---|---|---|
| 精製済み1st PCR産物 | ≧5ng | ≧0.2ng/μL | ≧20μL |
| サンプル | 総量 | 濃度 | 液量 |
|---|---|---|---|
| 精製済み2nd PCR産物 | ≧150ng | ≧5ng/μL | ≧20μL |
標準データ解析
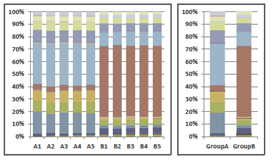
【菌叢構成情報の算出】
サンプルに含まれる菌の種類と占有率を算出します。
サンプルごと、群ごとの菌叢構成の傾向がわかります。
[菌叢構成のグラフ]
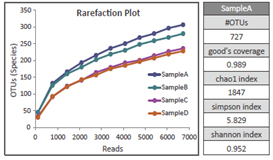
【α多様性情報の算出】
希薄化曲線の形状や群集の多様度を表す指標値により、菌叢の多様性の高さと取得データの充足度がわかります。
[α多様性情報]
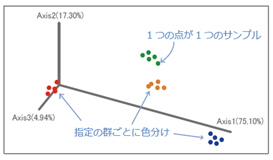
【PCoAプロット作成】
サンプル間の菌叢の類似度に基づき主座標分析を行います。PCoA プロットでは菌叢の似たサンプルが近くに集まり、群ごとのまとまりを視覚的に表すことができます。
[PCoAプロット]
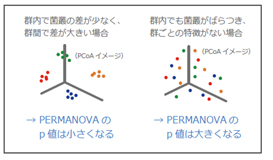
【有意差検定(PERMANOVA)】
PERMANOVA では、サンプルを指定の群に分けた時に群間で菌叢に有意な差があるかを調べることができます。PCoAプロットとセットで利用されることが多い解析です。
[PERMANOVA 解析イメージ]
オプションデータ解析
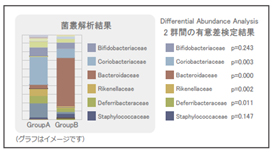
【有意差検定(Differential Abundance Analysis)】
群間における各菌の占有率の違いについて有意差検定を行います。p 値が低い菌は群間で占有率が有意に変動していると言えます。
[Differential Abundance Analysis 解析イメージ]
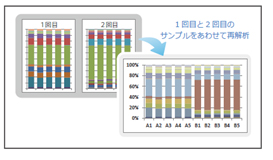
【複数データセットの統合】
複数回実施された群集解析について、比較対象のサンプルのデータを統合して再解析(上記、標準データ解析)を行います。
[複数データセットの統合]
よくある質問と回答
Q:解析に必要なリード数はどのくらいですか
サンプルにおける菌の多様度により、解析に十分なデータ量は異なりますが、弊社では標準的に10万リードペア/検体のデータ取得プランをご用意しています。データ解析時にα多様性情報を算出することで、データ量が十分であったかどうかを評価することができます。
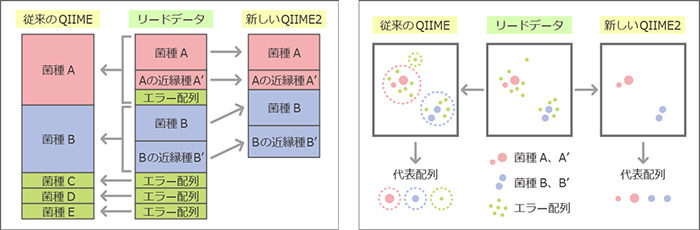
Q:解析プログラムQIIMEとQIIME2の違いは何ですか
旧解析プログラム「QIIME」では、相同性の高い配列をまとめて代表配列(OTU)を作成していたため、1つのOTUに複数の近縁種由来の配列が含まれる場合がありました。新しい「QIIME2」では、エラー配列の除去後、完全同一配列をまとめて代表配列とすることで、相同性の高い近縁種を分けて解析することができます。