- HOME
- Products & Services
- Application Selection Guide
- Isoform Sequencing (full-length mRNA-seq)
Isoform Sequencing (full-length mRNA-seq)
- Overview
- Analysis Principle
- Specifications
- Data Analysis
- Sample Requirements
- Frequently Asked Questions
Overview
This approach reads the entire mRNA molecule in a single pass, making it particularly effective for detailed analyses of splicing variants and discovery of novel isoforms, which are challenging with short-read sequencing.
With the latest updates to sequencing platforms and reagent kits, throughput has significantly improved, enabling not only qualitative but also quantitative data acquisition.
Analysis Principle
Consensus sequences are generated from the reads, representing the full-length transcript sequences.
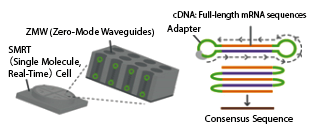
Specifications
| Platform | PacBio Revio |
| Reagent | Kinnex full-length RNA kit |
| Data Amount | 5 million HiFi reads/sample or 10 million HiFi reads/sample |
Data Analysis
・Raw Data Processing and Analysis
Generates full-length transcript sequences from raw reads using PacBio’s SMRT Link software.
・Alignment with Reference Genome
Aligns full-length transcript sequences to a specified reference genome.
・Functional Annotation of Transcripts
Predicts the functionality of obtained transcript sequences by performing similarity searches against established databases (e.g., NR, KOG/COG, Swiss-Prot).
・Gene Structure Analysis
Examines alternative splicing, alternative polyadenylation (APA), and novel gene/transcript sequences. Annotation of novel sequences is also performed.
・Gene/Transcript Expression Level Analysis
Calculates expression levels for genes and transcripts using the reference genome.
・Differential Gene/Transcript Expression Analysis
Evaluates expression variations between genes or transcripts based on expression level data.
・GO Enrichment Analysis (optional)
For analyses involving two or more groups, identifies enriched Gene Ontology (GO) terms for differentially expressed genes.
・Pathway Enrichment Analysis (optional)
For analyses involving two or more groups, identifies enriched pathways associated with differentially expressed genes.
Sample Requirements
| Sample Type | Total Amount | Concentration | Volume |
| Total RNA | ≥ 1.5 µg | ≥ 50 ng/µL | ≥ 30 µL |
Frequently Asked Questions
Q: Can I request analysis for a single sample?
Yes. Analyses are pooled on PacBio Revio, and you can choose between 5 million or 10 million HiFi reads starting from one sample.
Q: Can I customize the data amount?
Currently, only two scales―5 million or 10 million HiFi reads―are available. Customization is not supported.
Q: Are there recommended RNA extraction kits?
We do not specify a particular kit. We recommend using commercially available RNA extraction kits (e.g., QIAGEN RNeasy) along with DNase treatment.
Quality metrics: RIN ≥ 7, A260/280 between 1.8 and 2.0, A260/230 between 1.3 and 2.5.