- HOME
- 製品・サービス
- 目的別:アプリケーション選択ガイド
- ゲノムリシーケンス解析(ヒト以外)
-
核酸原薬CDMO
-
DNA・RNA・修飾核酸
-
次世代シーケンス(NGS)
- 次世代シーケンス(NGS)解析
-
目的別:アプリケーション選択ガイド
- 目的別:アプリケーション選択ガイド
- ヒトゲノム解析
- ゲノムリシーケンス解析(ヒト以外)
- 微生物ゲノム配列決定
- 小スケール解析(NGS Petit)
- GRAS-Di®-ジェノタイピングシーケンス解析
- リピートモチーフ検索
- 一本鎖・微量・損傷 DNA対応ライブラリ調製
- 遺伝子発現解析(リファレンス配列のある生物)
- Iso-Seq 解析(full-length mRNA-seq)
- De novoトランスクリプトーム解析
- Small RNA-Seq解析
- 微生物群集解析
- ショットガンメタゲノム解析
- メタトランスクリプトーム解析
- ChIP-Seq解析
- CRISPRスクリーニング解析
- Illuminaアンプリコンシーケンス解析
- PacBioアンプリコンシーケンス解析
- 標準パイプライン解析
- カスタム解析
- 病理組織標本から遺伝子解析
- シーケンサー機種・原理
- サンプル必要量
- ご注文方法
- NGS論文実績
-
DNAシーケンス
-
マイクロアレイ
-
タンパク質関連
-
関連製品・サービス
ゲノムリシーケンス解析(ヒト以外)
解析概要
ゲノムシーケンスを行い、ゲノム上の変異候補箇所を検索します。変異株の原因遺伝子を探索することができます。
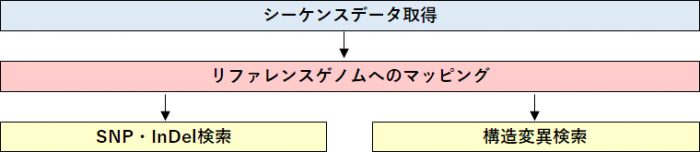
解析の流れ
推奨データ取得量
| シーケンス仕様 | Illumina 150PE |
| データ量 | ゲノムサイズの×30以上 ※詳細はQ&Aをご参照ください 例)シロイヌナズナ:ゲノムサイズ130Mb → データ量4Gb以上推奨 |
解析メニュー
【マッピング・SNP候補検索】
リファレンスゲノムにリードをマッピングし、リファレンス配列とリード配列を比較してSNP候補箇所を検索します。
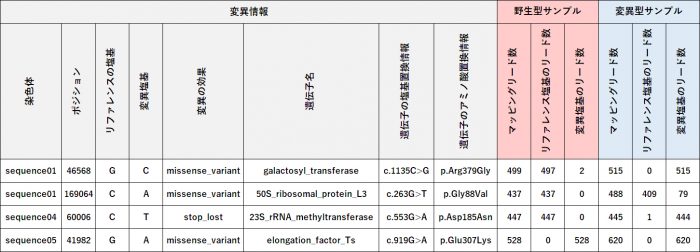
納品データ例:変異検索結果(EXCEL形式)
【マッピング・構造変異検索】
リファレンスゲノムにリードをマッピングし、ペアリードのマッピング状況にもとづき、大規模な構造変異(挿入・欠失・逆位・転座)を検索します。
サンプル必要量
| ライブラリ調製方法 | 提供サンプル種別 | 総量 | 濃度 | 液量 |
| PCR-Plus | DNA | 500 ng 以上 | 10 ng/uL 以上 | 30 uL 以上 |
| PCR-Free | 4 ug 以上 | 50 ng/uL 以上 | 30 uL 以上 |
よくある質問と回答
Q:カバレッジと取得データ量
カバレッジは、ゲノム上の任意のポジションをカバーするリードの平均本数に相当し、「取得データ量(bp)÷ ゲノムサイズ(bp)」で算出されます。
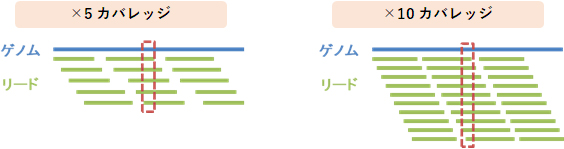
例えば平均×30カバレッジのデータで変異解析を行った場合、ホモの変異は30本のリードのほぼ全てで検出され、ヘテロの変異は30本中15本程度のリードで検出されます。カバレッジは領域により多少増減するため、最低でも平均×30カバレッジ以上のデータ量を取得していただくことを推奨いたします。
Q:コントロールサンプルの必要性
変異株1検体のみで変異検索を行うと、リファレンスゲノムとサンプルの個体差や、ミスマッピングに由来する変異候補も多数検出されます。コントロールサンプルとして、親株や野生株の解析を行うことで、そのようなバックグラウンドに相当する変異候補箇所を除外することができます。
Q:バルクサンプルでの解析
真菌や植物において変異原処理により得られた変異体では、形質と無関係の変異も多く生じています。戻し交配で得られた変異株10~30個体のバルクサンプルを使用して解析を行うことで、変異頻度に基づき原因変異とそれ以外の変異を区別することができます。その際、野生型のバルクサンプルや、親株のサンプルをコントロールとして使用いたします。(上記「コントロールサンプルの必要性」参照)
Q:微生物ゲノムの解析
ゲノムサイズの小さい微生物では、公共データベースのリファレンスゲノムとサンプルの系統が離れている場合、野生株サンプルのデータを使用してリファレンスゲノムを作成することをお勧めします。(微生物ゲノム配列決定参照)
Q:RNA-Seqデータを使用して変異検索を行うことはできますか
RNA-Seq データを使用して、ある程度発現している遺伝子についてはSNPを検索することができます。ただし、低発現遺伝子やスプライスジャンクション付近において偽陽性の変異が検出されやすい、対立遺伝子間で発現量が異なる場合にゲノム上の変異頻度を正しく算出できないといったデメリットがあります。