-
核酸医薬品原薬CRDMO
-
DNA・RNA・修飾核酸
-
次世代シーケンス(NGS)
- 次世代シーケンス(NGS)解析
-
目的別:アプリケーション選択ガイド
- 目的別:アプリケーション選択ガイド
- ヒトゲノム解析
- ゲノムリシーケンス解析(ヒト以外)
- 微生物ゲノム配列決定
- 小スケール解析(NGS Petit)
- GRAS-Di®-ジェノタイピングシーケンス解析
- リピートモチーフ検索
- 一本鎖・微量・損傷 DNA対応ライブラリ調製
- 遺伝子発現解析(リファレンス配列のある生物)
- Iso-Seq 解析(full-length mRNA-seq)
- De novoトランスクリプトーム解析
- Small RNA-Seq解析
- 微生物群集解析
- ショットガンメタゲノム解析
- メタトランスクリプトーム解析
- ChIP-Seq解析
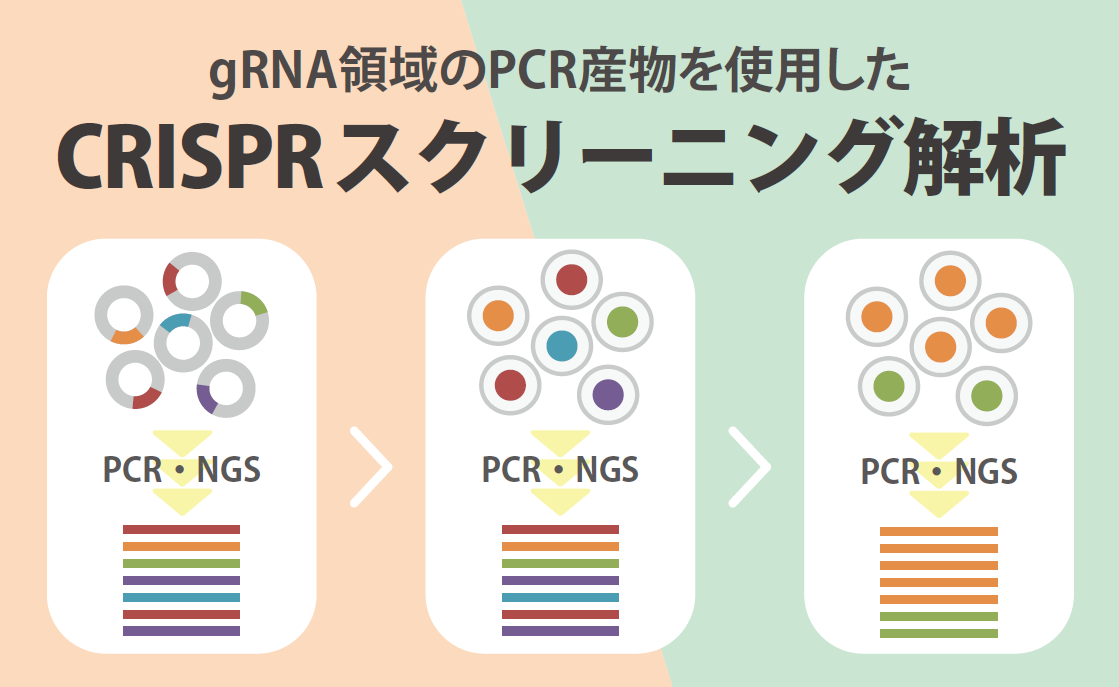
- CRISPRスクリーニング解析
- Illuminaアンプリコンシーケンス解析
- PacBioアンプリコンシーケンス解析
- 標準パイプライン解析
- カスタム解析
- 病理組織標本から遺伝子解析
- シーケンサー機種・原理
- サンプル必要量
- ご注文方法
- NGS論文実績
-
DNAシーケンス
-
マイクロアレイ
-
タンパク質関連
-
関連製品・サービス
次世代シーケンス(NGS)解析
北海道システム・サイエンス株式会社では、次世代シーケンス(NGS)受託解析サービスを承ります。
お客様の解析目的に応じて、最適な解析プランをご提案します。まずはご相談ください。
次世代シーケンス(NGS)解析
詳細は以下のリンクよりご確認ください。
ご研究の目的別に、次世代シーケンス解析サービスメニューをご紹介しています。
#更新情報
・ WGS/WESキャンペーン開催(2025/08)
・ ISO-Seq 解析リニューアル(2024/12)
・ 年度末早割キャンペーン開催 (RNA-Seq / small RNA-Seq / CRISPR)(2024/11)
・ 核酸医薬品 オフターゲット検索/評価サービスを追加(2024/06)
・ CRISPRスクリーニング解析サービスのHP・注文書を更新(2024/05)

※ 新型コロナウイルス・インフルエンザウイルスを含むサンプルはお受入れできません。
弊社では【SARS-CoV-2】および【Influenza A/B virus】のPCR検査用オリゴを製造しているため、鋳型となり得る核酸サンプルの受け入れを中止しています。何卒ご了承ください。
お役立ち情報
弊社ではお客様のお困りごと・問題解決に向けたご提案を通じて、ご研究のサポートを行っています。
お役立ち情報ページでは、弊社で実施した解析事例・課題解決事例などをご紹介しています。
〇 解決事例

実験相談窓口
次世代シーケンス解析が初めての方から、既に取得済みのデータをお持ちの方まで、お客様の目的に合わせた最適なプランをご提案しています。
実験相談窓口をご用意してお待ちしていますので、どんなお悩みでも、まずはお気軽にご相談ください。


※ 「実験相談窓口」ページへのリンクはこちら
※ 「実験相談窓口」お問い合わせフォームへのリンクはこちら
NGSおすすめサービス
| CRISPRスクリーニング解析 | 微生物ゲノム配列決定 | 微生物群集解 |
 |
 |
 |
| Illuminaアンプリコンシーケンス解析 | 小スケール解析(NGS Petit) | カスタム解析 |
 |
 |
 |