- HOME
- 製品・サービス
- 目的別:アプリケーション選択ガイド
- De novoトランスクリプトーム解析
De novoトランスクリプトーム解析
解析概要
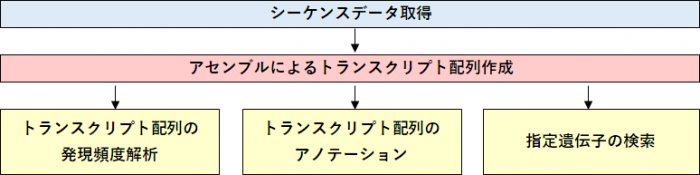
RNA-Seqデータからトランスクリプト配列を構築します。リファレンスゲノムの無い生物で、遺伝子検索や発現解析を行うことができます。
解析の流れ
推奨データ取得量
| シーケンス仕様 | Illumina 150PE |
| データ量 | 2,000万リードペア/検体 ※詳細はQ&Aをご参照ください |
解析メニュー
【De novoアセンブル】
リードのアセンブルを行い、トランスクリプト配列を構築します。複数サンプルの混合アセンブルについては、Q&Aをご参照ください。
納品データ例:トランスクリプト配列(FASTA形式)
ATTTTTATTTTGATGTGAAAAGTTAATTTTTTAACTATAAAATATTTTGCTTCAACAAGA
CAATATCCGCCAAATTAAATGAATATGCATTTATTATCTTTACCTGATTGTATGCTGGAA
…>TR1|c0_g1_i2
CGCCGAGAATCCGAGAGGAAAAAATGTATTTTCTTTCAGAAATCATCCACTTGCGAAACA
TGCTGACATACTAACTTGTATTGAAACAAGAATATCCATGCTTTCAATGACATACACCAA
…
【アノテーション情報付与】
アセンブルで構築したトランスクリプト配列に対して、既知遺伝子データベースを参照したBLAST検索を行い、相同遺伝子情報を付与します。
【マッピング・発現頻度解析】
アセンブルで構築したトランスクリプト配列に対して、元のリードをマッピングし、遺伝子発現量を算出します。サンプル間・グループ間の比較も可能です。
納品データ例:発現頻度解析結果(EXCEL形式)
| gene_id | Sample A | Sample B | Sample B | |||
| Sample A | ||||||
| FPKM | FPKM | logFC | logCPM | PValue | FDR | |
| TR10000|c0_g1 | 0.49 | 0.13 | -1.824253682 | -2.050135117 | 0.430641822 | 1 |
| TR10001|c0_g1 | 0.13 | 0.7 | 2.238008044 | -1.894306859 | 0.282608696 | 0.908307924 |
| TR10002|c0_g1 | 0.58 | 0.25 | -1.218299202 | -1.760545939 | 0.52173913 | 1 |
| TR10003|c0_g1 | 0.08 | 0.77 | 3.070569548 | -1.407003958 | 0.057771114 | 0.475628079 |
| TR10004|c0_g1 | 0.5 | 0.77 | 0.52824399 | -1.217384592 | 0.82249198 | 1 |
| TR10005|c0_g1 | 20.61 | 16.37 | -0.408638264 | 3.253900296 | 0.555121307 | 1 |
| … | … | … | … | … | … | … |
【指定配列の検索】
アセンブルで構築したトランスクリプト配列の中から、指定配列※に相同な配列を検索します。
※ 既知のペプチド断片配列や、近縁種の遺伝子など
サンプル必要量
| 生物種 | 提供サンプル種別 | 総量 | 濃度 | 液量 |
| 真核生物 | total RNA | 600 ng 以上 | 20 ng/uL 以上 | 30 uL 以上 |
| mRNA | 100 ng 以上 | 3 ng/uL 以上 | 30 uL 以上 | |
| 原核生物 | total RNA | 1.5 ug 以上 | 50 ng/uL 以上 | 30 uL 以上 |
| mRNA | 100 ng 以上 | 3 ng/uL 以上 | 30 uL 以上 |
よくある質問と回答
Q:必要データ量
1検体のみでアセンブルを行う場合は、5,000万リードペア/検体以上のデータ量を推奨致します。複数検体のデータを混合してアセンブルを行う場合は、1,000万~2,000万リードペア/検体程度から解析可能です。データ量を増加すると、より低発現の遺伝子情報を得ることができます。
Q:de novoアセンブルで得られるコンティグの数や長さ
生物種やシーケンスデータ量にもよりますが、平均800~1,000bp程度、10万本~30万本程度のコンティグが得られます。転写産物の全長配列を取得することを目的とする場合は、PacBioシーケンサーを使用したIso-Seq解析をご案内いたします。
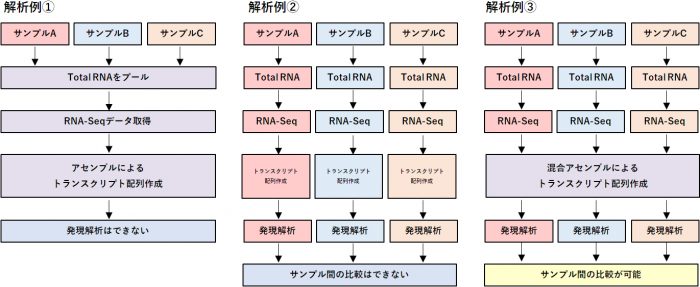
Q:混合アセンブルについて
生物の遺伝子配列情報を取得すること自体を目的とした解析では、さまざまな組織のTotal RNA をプールし、遺伝子発現プロファイルが均衡化されたサンプルでシーケンス、アセンブル解析を行います(①)。各検体の発現プロファイルも取得したい場合(②)、検体間の発現比較解析を行いたい場合(③)は、検体ごとにRNA-Seq データを取得する必要があります。
Q:発現解析のレプリケートについて
発現変動の有意差検定を行うためにはn=3以上のレプリケートをご用意いただくことを推奨しています。レプリケートがない場合でも、検体間の発現比情報(Fold Change)を得ることはできます。
Q:バクテリアでもDenovoトランスクリプトーム解析が可能ですか
バクテリアの場合はDenovoトランスクリプトーム解析ではなく、ゲノムシーケンスによるゲノム構築を行い、ゲノム上の遺伝子予測により遺伝子情報を取得していただくことをお勧めします。