- HOME
- 製品・サービス
- 目的別:アプリケーション選択ガイド
- Iso-Seq 解析(full-length mRNA-seq)
Iso-Seq 解析(full-length mRNA-seq)
解析概要
PacBioシーケンサーを使用したロングリード技術による網羅的な全長RNA解析です。
mRNA分子を1リードで読み切ることが可能なため、ショートリード解析では困難だったスプライシングバリアントの詳細な解析や、新規アイソフォームの探索などに最適です。
シーケンス機器、試薬キットのアップデートにより、スループットが大幅に向上し従来の定性的な解析に加え、定量的な情報の取得も可能になりました。
- ※ ライブラリ作製では Oligo dT を使用した cDNA 合成が必要なため、解析対象は polyA RNA に限定されます。
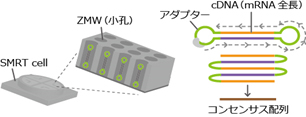
解析原理
mRNAを断片化せずにライブラリ化し、SMRT Cellの小孔で1分子のcDNAを繰り返しシーケンスします。 リードのコンセンサス配列が、トランスクリプトの 全長配列となります。
解析仕様
| 機種 | PacBio Revio |
| 試薬キット | Kinnex full-length RNA kit |
| データ量 | 500万 HiFi reads/sample または 1000万 HiFi reads/sample |
- ※ 500万 HiFi reads または 1000万 HiFi reads の2つのスケールからお選びいただけます。Revioの乗り合い解析により、最低 1サンプル からご利用いただけます。
解析メニュー
・Raw Data Processing and Analysis
PacBioシーケンサー付属のSMRT-Linkにより、得られたRawリードから全長トランスクリプト配列を生成します。
・Alignment with Reference Genome
参照ゲノム配列を指定し、取得した全長トランスクリプト配列をアライメントします。
・Functional Annotation of Transcripts
得られたトランスクリプト配列がどのような機能をもつか推定します。既存のデータベース (NR, KOG/COG, Swiss-prot…等)内の配列との相同性検索を行い配列の類似性を評価します。
・Gene Structure Analysis
得られたトランスクリプト配列情報を基に、Alternative splicing、Alternative polyadenylation (APA)、新規遺伝子/トランスクリプト配列の解析を行い、新規配列へのアノテーションも行います。
・Gene/Transcript Expression Level Analysis
参照ゲノム配列を利用し、遺伝子/トランスクリプト単位で発現量を算出します。
・Differential Gene/Transcript Expression Analysis
遺伝子/トランスクリプトの発現量情報を基に、発現変動の有無について評価を行います。
・GO Enrichment Analysis (for two or more groups)
2群以上のグループで解析を行った場合、グループ間で発現変動の見られた遺伝子群についてGOエンリッチメント解析を行います。
・Pathway Enrichment Analysis (for two or more groups)
2群以上のグループで解析を行った場合、グループ間で発現変動の見られた遺伝子群についてPathwayエンリッチメント解析を行います。
サンプル必要量
| 提供サンプル種別 | 総量 | 濃度 | 液量 |
| total RNA | 1.5 µg 以上 | 50 ng/µL 以上 | 30 µL 以上 |
よくある質問と回答
Q:1サンプルでも依頼できますか?
PacBio Revioによる乗り合い解析での解析になりますので、500万/1000万 HiFi readsどちらのスケールでも1サンプルからご利用いただけます。
Q:取得するデータ量は任意で変更できますか?
現在、500万または1000万 HiFi reads の2パターンのみ対応しており、スケールの任意変更はできません。ご了承ください。
Q:RNAを抽出する際に推奨されるキット等はありますか?
推奨品はございません。市販のRNA抽出キット(QIAGEN RNeasy 等)をご使用いただき、DNase処理も併せて実施されることをおすすめしております。
RIN値 7以上、A260/280 1.8 – 2.0、A260/230 1.3 – 2.5 が純度の目安となります。